We support biopharma companies which need to effectively disseminate key information to the industry stakeholders including regulators, patients, and healthcare professionals, and bioinformatics companies which need to navigate the complex regulatory frameworks. We provide standardized formats tailored as per good clinical practices (ICH-GCP) and other regulatory guidelines and policies, and provide bespoke tools to adapt materials to commonly used frameworks, such as the NHS Digital Technology Assessment Criteria (DTAC).
Common Technical Documents. The following table provides a list of the most common pre-approval regulatory documents for drugs that we can provide and their associated guidelines – regulations in which we support our work.

Clinical study reports (CSRs). We have created CSRs as part of the process of submitting applications for new medical treatments to regulators. Our CSRs aimed to answer the questions such as: Why was the trial done? What were the important questions asked in the trial? What were the results? We also included extensive details on the course of treatment for the patients under the therapy, the medical information collected from the patients as part of the research, and demographic data. Another type of information provided in our CSRs was based on how the trial was conducted and how the results were analyzed.
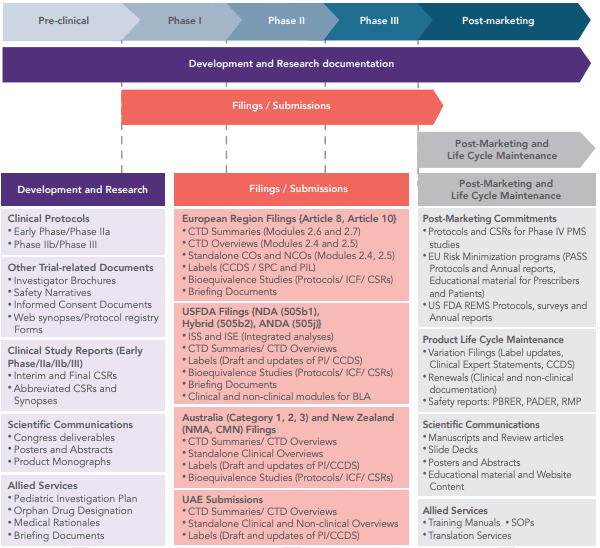
Clinical trial protocols. We provide the needed support for a clear trial design to meet the study objectives. We help to understand and be ready to review and discuss aspects of the trial design with the protocol development team, in order to write a clear and accurate protocol.
